Performance and Proficiency
As IRIDA instances are locally installed (as opposed to a web-based service), the speed and scalability of the software will largely depend on local computing power and infrastructure.
Performance has been evaluated for the following IRIDA software components:
SNVPhyl
Benchmarking of IRIDA’s assembly and variant calling workflow has previously been demonstrated to be robust, and repeatedly performs above average compared to other de novo assembly and variant callers (Petkau et al, 2017).
IRIDA’s SNVPhyl (Single Variant Nucleotide Phylogenomics) pipeline quickly analyzes many genomes, identifies variants, generates maximum-likelihood phylogenies and all-against-all SNV distance matrices, and additional quality information to help guide interpretation of the results. Through the use of Galaxy, SNVPhyl is able to integrate with most major higher performance computer scheduling engines to independently distribute the workload for each genome across a cluster as well as fine-tuning resource requirements (e.g., memory or CPU cores) for each individual stage of the workflow.
SNVPhyl has been used for hundreds of public health analyses at Canada’s National Microbiology Laboratory (NML, Public Health Agency of Canada) and to support research and provincial laboratory services since 2010. The pipeline is currently being used for outbreak investigations and has been validated as part of a suite of tools used by PulseNet Canada for all genomic epidemiological investigations of foodborne outbreaks since 2012.
At the NML, variant detection of a single genome takes ~30mins; however, it can be scaled to the simultaneous analysis of thousands of genomes in a similar timeframe. SNVPhyl can produce a phylogeny of 100 isolates in ~1 hour. Furthermore, SNVPhyl detects SNVs in both simulated and real-world data with a high degree of sensitivity and specificity, rapidly removes SNVs within regions of homologous recombination, and correctly distinguishes outbreak-related isolates from non-outbreak isolates across a range of parameters and sequencing data qualities (Petkau et al, 2017).
IRIDA’s SNVPhyl pipeline also has been involved in different international proficiency testing exercises performed by various organizations such as the Global Microbial Identifier and the American Society for Microbiology. Such competitions provide opportunities to compare and contrast software performance and analysis methods for classifying isolates from a real-world outbreak scenarios using next-generation sequencing data. In these exercises, SNVPhyl analyses demonstrated consistency with other pipelines. More in-depth comparisons of SNVPhyl with other pipelines (Katz et al, 2017; Usonga et al, 2018) demonstrated 100% concordance in terms of clustering isolates into outbreaks, and distinguishing non-outbreak isolates, across a variety of organisms.
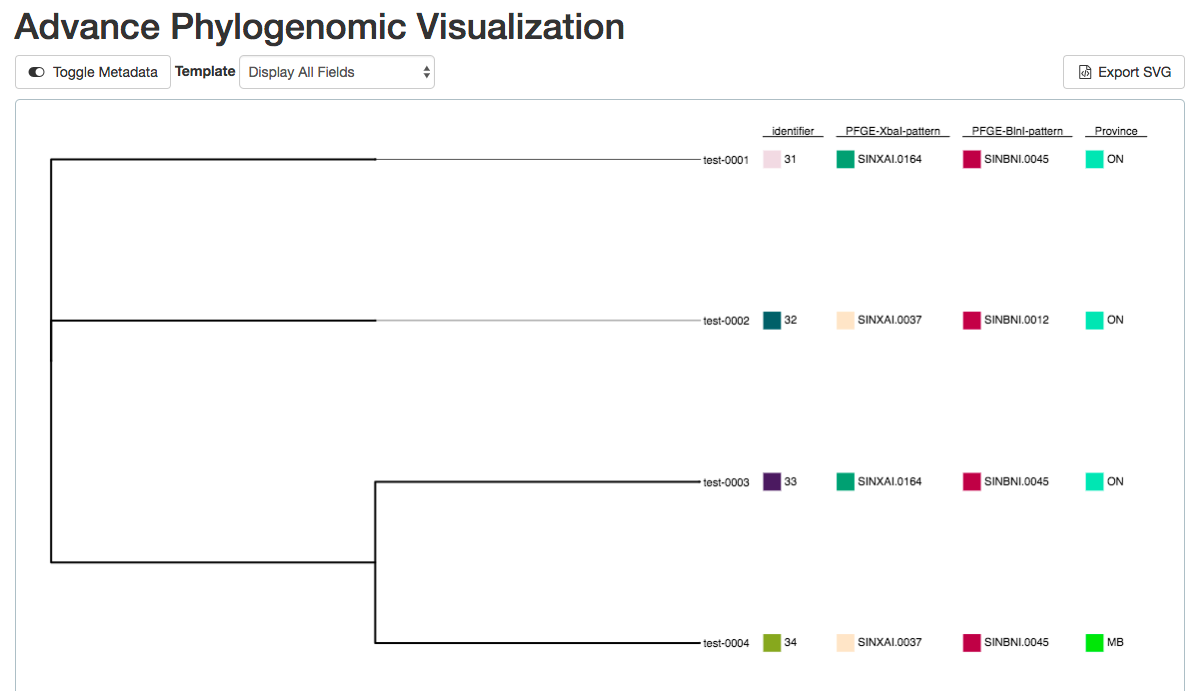 IRIDA’s advanced visualization of SNVPhyl’s phylogenomic analyses enables integration and dynamic filtering of user-defined metadata, to better label and characterize clusters for outbreak investigation and epidemiological decision-making.
IRIDA’s advanced visualization of SNVPhyl’s phylogenomic analyses enables integration and dynamic filtering of user-defined metadata, to better label and characterize clusters for outbreak investigation and epidemiological decision-making.
The Salmonella In Silico Typing Resource (SISTR)
Due to the critical importance of Salmonella serovar information to public health, it is essential to produce fast and reliable serovar assignments for surveillance and investigations. Current serotyping tests generate one result per assay, however WGS is increasingly being utilized as a single diagnostic test. Whole genome data is capable of replacing a number of costly and labour intensive assays. SISTR has also been extensively validated, yielding accuracies of ~95%, the highest among serotype prediction tools (Yoshida et al, 2016). SISTR is currently being used to generate serotype predictions for all genomes submitted to EnteroBase, the largest repository of Salmonella WGS data worldwide. SISTR was also recently used to generate predictions for the 30% of Salmonella genomes that have been deposited at NCBI with missing serovar information. Furthermore, this study provided a large validated set of genomes, which can be used to benchmark new bioinformatics tools (Robertson et al, 2018). Implementation of SISTR has also led to the phasing out of antigen-based serotyping at Canada’s National Microbiology Laboratory, which has moved to WGS as the primary means of characterization of Salmonella isolates from national surveillance programs as of May 2017 (Yachison et al, 2017). SISTR is suitable for generating in silico serovar nomenclature compatible with historical records, surveillance systems, and communication structures currently in place.
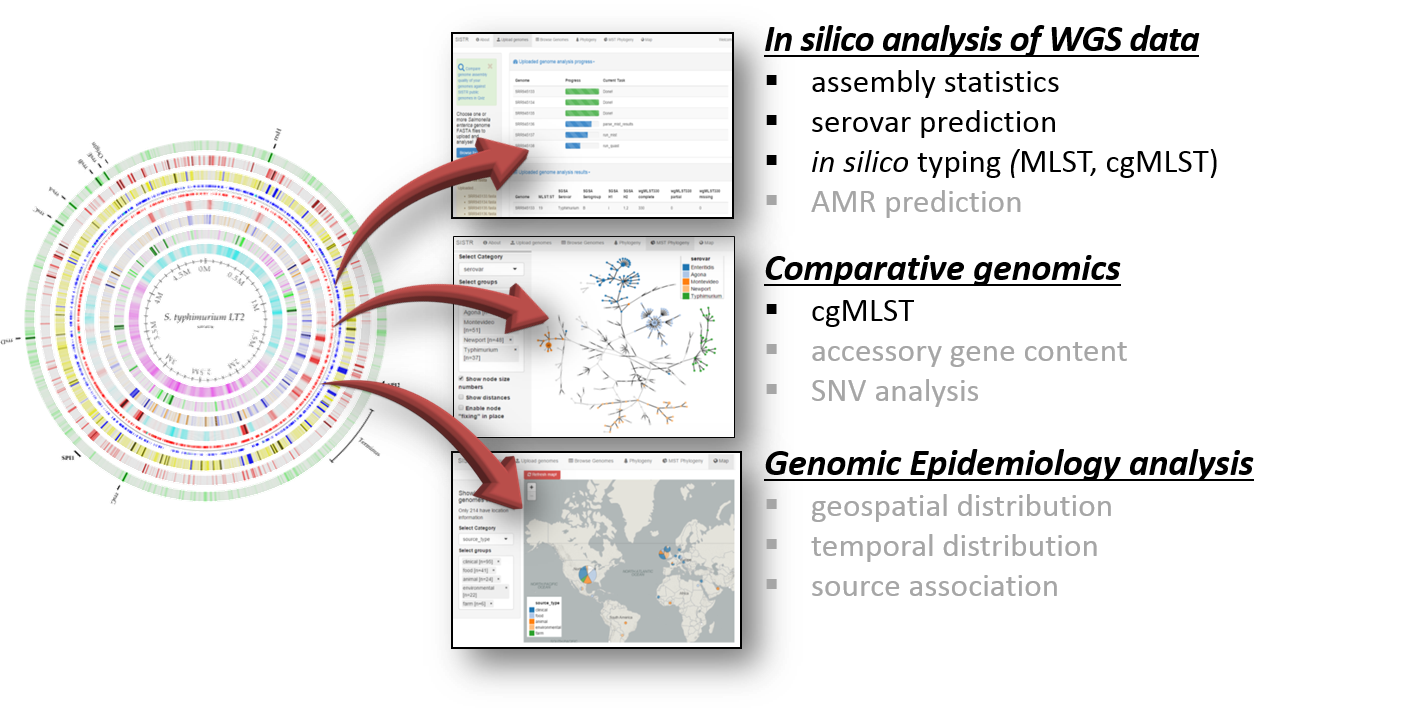 SISTR enables Salmonella genome assembly, in silico serovar prediction, as well as cgMLST and MLST.
SISTR enables Salmonella genome assembly, in silico serovar prediction, as well as cgMLST and MLST.
MentaLiST
MentaLiST is an MLST and cgMLST calculation engine that generates an ‘allelic profile’ based on an established MLST schema. MentaLiST has been demonstrated to be faster than other MLST callers while providing the same or better accuracy (Feijao et al, 2018), requiring on average ~30s per sample to run. Furthermore, MentaLiST is capable of dealing with MLST schemes with up to thousands of genes while requiring limited computational resources.